Interestingly, the hypothalamus isn’t the only place where SS is contained; the thyroid gland also contains Somatostatin-producing cells. This is of interest to us, because in the case of the thyroid, it’s been noted that certain hormones which were previously thought only to govern GH secretion can also influence thyroid hormone output as well. SS can directly act to inhibit TSH secretion or it may act on the hypothalamus to inhibit TRHsecretion. So when you add GH into your body from an outside source, you are triggering the body into releasing SS, because your body no longer needs to produce its own supply of GH…and unfortunately, the release of SS can also inhibit TSH, and therefore limit the amount of T4 your body produces.
In addition, as IGF-I production isincreased in the hypothalamus after T3 administration and T3 may participate in IGF-1 mediated negative feedback of GH by triggeringeither increased somatostatin tone and/or decreased GHRH production (6). IGF, interestingly, has the ability to mediate some of T3’s effects independent of GH, but not to the same degree GH can (7.) In fact, IGF-I production isincreased in the hypothalamus after T3, administration it may plausibly participate in negative feedback by triggeringeither increased somatostatin tone and/or decreased GHRH production.So we know that GH lowers T4 (more about this in a sec), but an increase in T3 upregulates GH receptors (8) as well as IGF-1 receptors (9,10).
As can be previously stated, and due to the ability of GH to convert inactive T4 into active T3, GH administration in healthy athletes shows us an entirely predicatble increase in mean free T3 (fT3), and a decrease in mean free T4 (fT4)levels.(11)
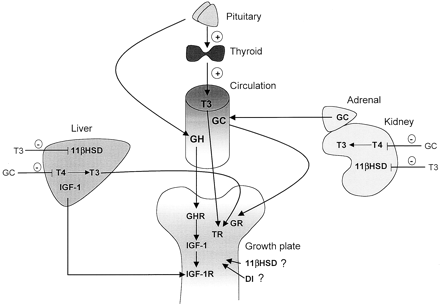
As you can see, T3 levels are directly correlative to GH gene transcription. The scientists who conducted the study which provided the graph above concluded that the amount of T3 present is a regulatory factor on how much GH gene transcription actually occurs. And gene transcription is what actually gives us the effects from GH. This last fact really seems to shed some light on why we need T3 levels to be supraphysiological if we’re going to be using supraphysiological levels of GH, right? Otherwise, the GH we’re using is going to be limited by the amount of T3 our body produces. However, since we’re taking GH, and it is converting more T4 into T3, T4 levels are lowered substantially, and this is the problem with GH. and may actually be THE limiting factor on GH…if we assume that at least some of GH’s effects are enhanced by thyroid hormone, and specifically T3, then what we are looking at is the GH that has been injected is being limited by a lack of T3. But that doesn’t make sense, because if we use T3 + GH, we get a decrease in the anabolic effect of GH.
Additional T3 is not all that’s needed here. What’s needed is the actual conversion process of T4-T3, and the deiodinase presence and activity that it involves. This is because Local 5′-deiodination of l-thyroxine (T4) to active the thyroid hormone 3,3′,5-tri-iodothyronine (T3) is catalyzed by the two 5′-deiodinase enzymes (D1 and D2). These enzymes not only “create” T3 out of T4, but actually regulates various T(3)-dependent functions in many tissues including the anterior pituitary and liver. So when there is an excess of T3 in the body, but normal levels of T4, the body’s thyroid axis sends a negative feedback signal., and produces less (D1 and D2) deiodinase, but more of the D3 type, which signals the cessation of the T4-T3 conversion process, and is inhibitory of many of the synergistic effects that T3 has! Remember, Type 3 iodothyronine deiodinase (D3) is the physiologic INACTIVATOR of thyroid hormones and their effects (13)and is well known to have independent interaction with growth factors (which is what GH and IGF-1 are).(14) This is because with adequate T4 and excess T3, (D1 and D2) deiodinase is no longer needed for conversion of T4 into T3, but levels of D3 deiodinase will be elevated. When there is less of the first two types of deidinase, it would seem that the T3 which has been converted to T4 can not exert it’s protein sparing (anabolic effects), as those first two types are responsible for mediation of many of the effects T3 has on the body. This seems to be one of the ways deiodinase contributes to anabolism in the presence of other hormones.
In other words, if we have enough to GH in our body aid in supraphysiological conversion of T4 into T3, but we already have the too much (exogenous) T3, the GH is not going to be converting any excess T4 into T3 after a certain point- which would be a limiting factor in GH’s anabolic effects, when coupled with the act that we’ve allowed the D3 enzyme to inhibit the T3/GH synergy that is necessary.
So what are we doing when we add T3 to GH? We’re effectively shutting down the conversion pathway that is responsible for some of GH’s effects! And what would we be doing if we added in T4 instead of T3? You got it- we’d be enhancing the pathway by allowing the GH we’re using to have more T4 to convert to T3, thus giving us more of an effect from the GH we’re taking. Adding T4 into our GH cycles will actually allow more of the GH to be used effectively!
So we want elevated T3 levels when we take GH, or we won’t be getting ANYWHERE NEAR the full anabolic effect of our injectable GH without enough T3. And now we know that not only do we need the additional T3, but we actually want the CONVERSION process of T4 into T3 to take place, because it’s the presence of those mediator enzymes that will allow the T3 to be synergistic with GH, instead of being inhibitory as is seen when T3 is simply added to a GH cycle. And remember, we don’t only want T3 levels high, but we want types 1 and 2 deiodinase to get us there- and when we take supplemental T3, that just doesn’t happen…all that happens is the type 3 deiodinase enzyme shows up and negates the beneficial effects of the T3 when we combine it with GH.





 Reply With Quote
Reply With Quote
